The GC content is the percentage of guanine (G) and cytosine (C) in a DNA or RNA sequence. This measure is one of the metrics which can be used in the sequencing quality control (e.g., detects contamination).
Here we will use a small example to see how this metric is calculated.
# Generate a list of DNA sequencesdna_sequences <-list(seq1 ="ATGCGTAGCTAGGCTATCCGA",seq2 ="CGCGTTAGGCAAGTGTTACG",seq3 ="GGTACGATCGATGCGCGTAA",seq4 ="TTTAAACCCGGGATATAAAA")# the 1st DNA sequencedna_sequences[["seq1"]]
[1] "ATGCGTAGCTAGGCTATCCGA"
Split the 1st DNA sequence into individual bases using the function strsplit(). (See ?strsplit)
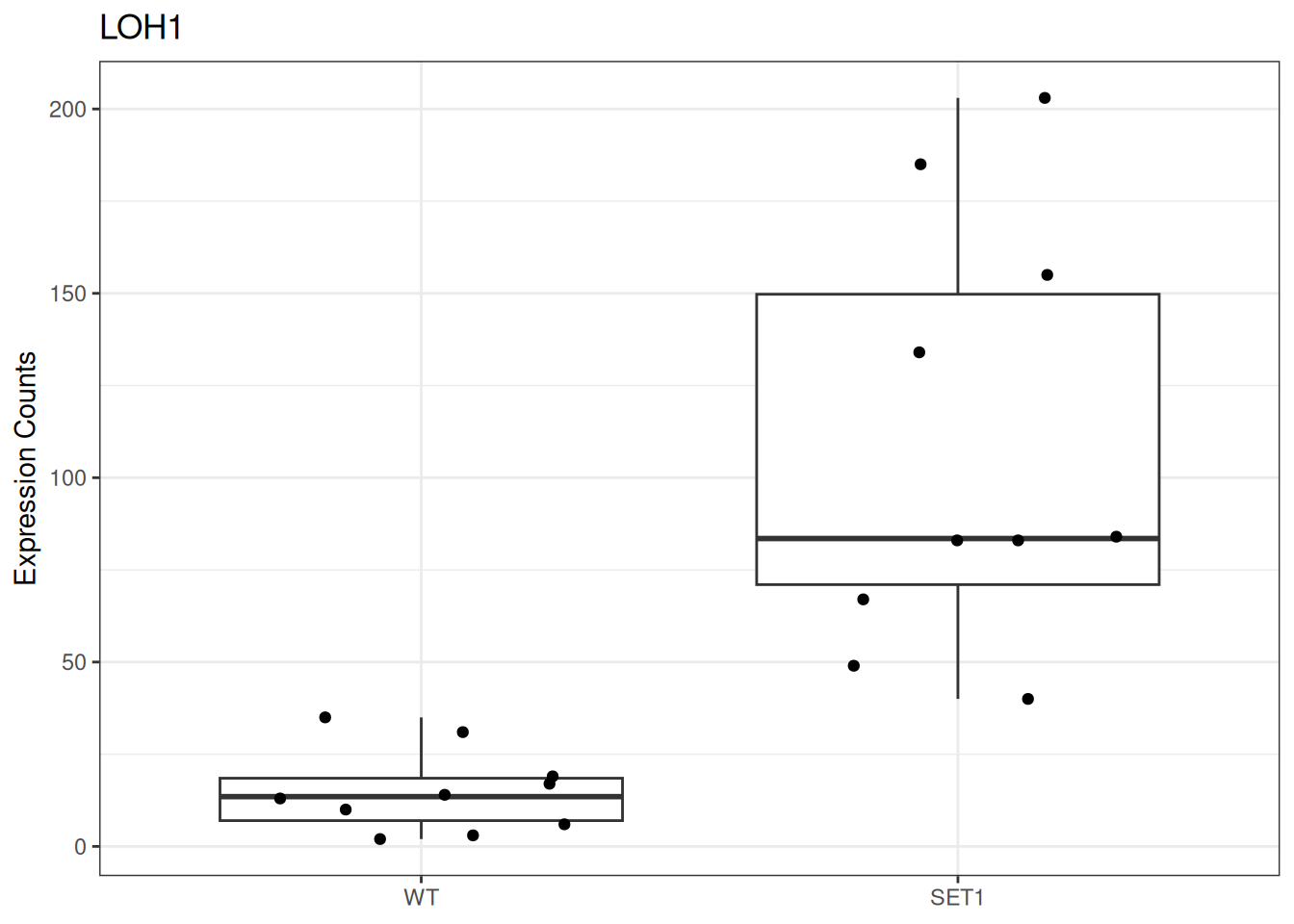
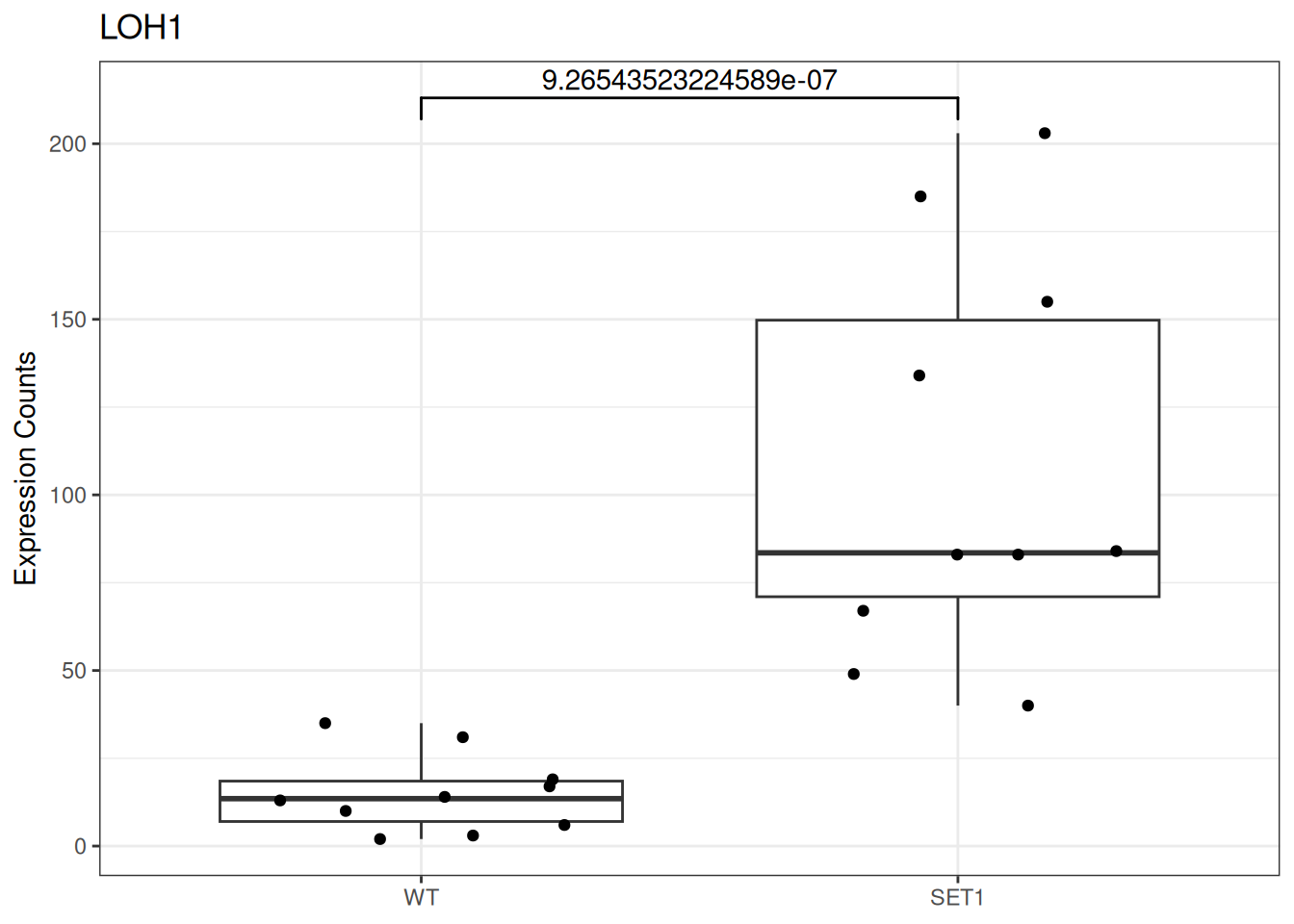
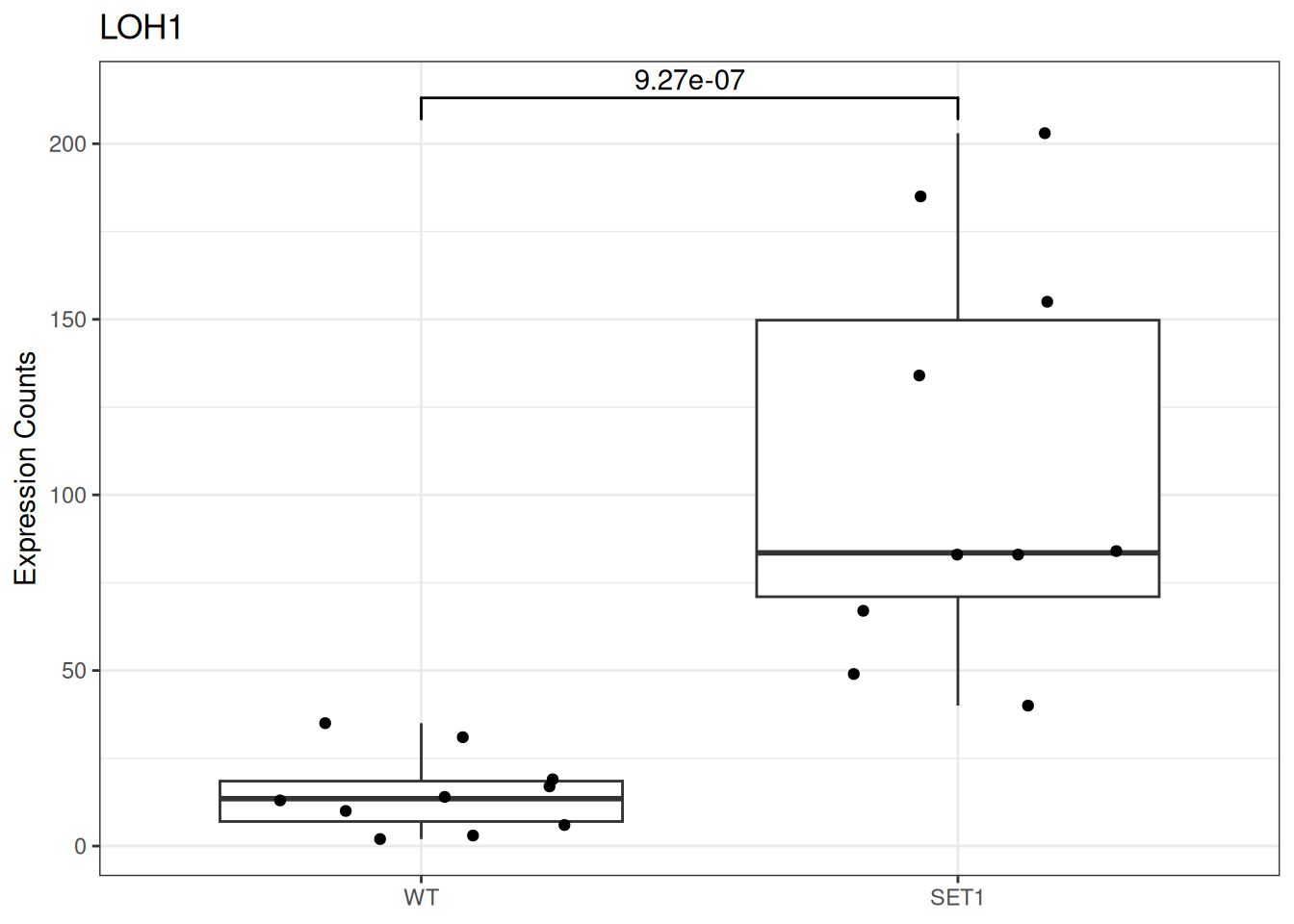
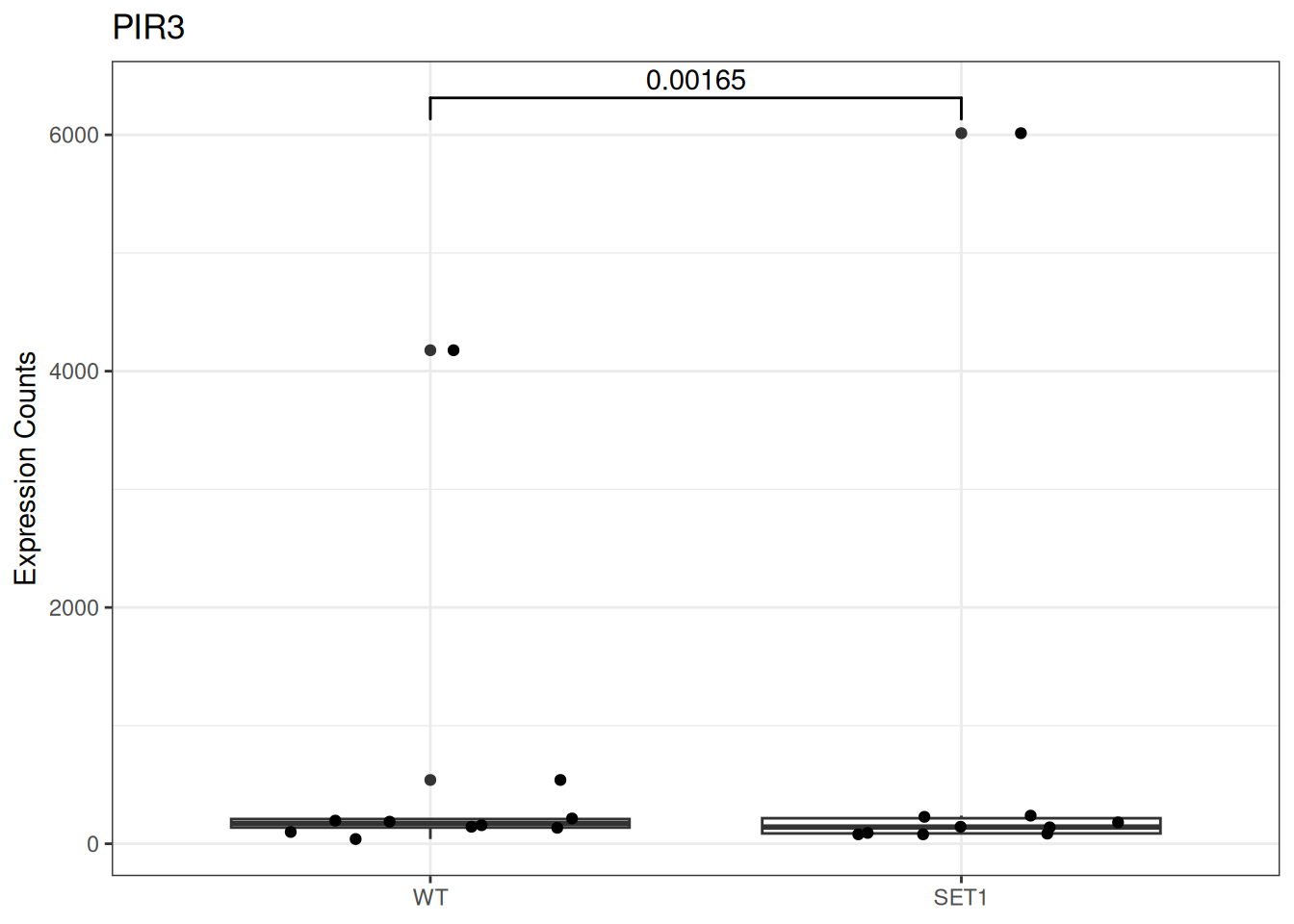
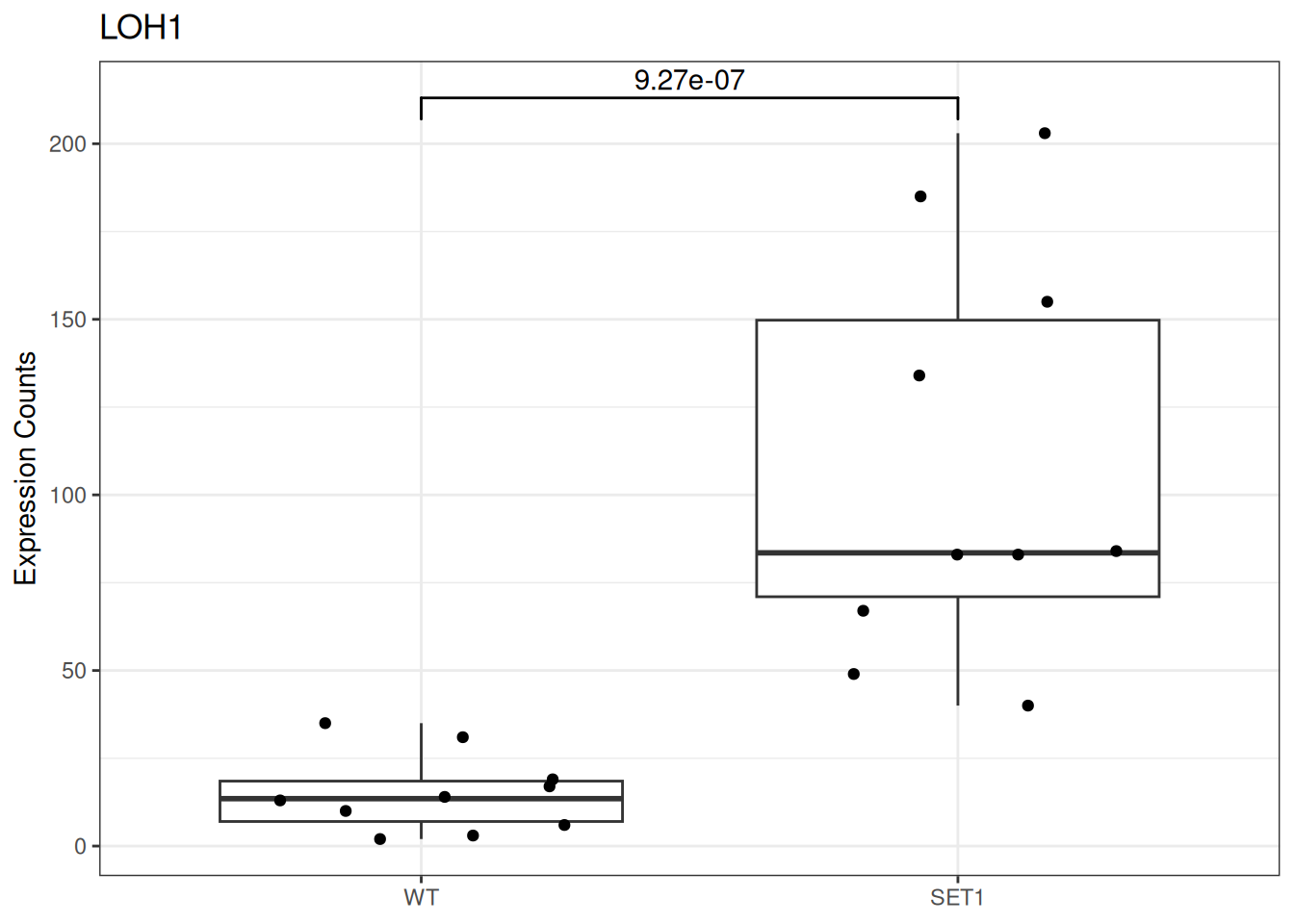
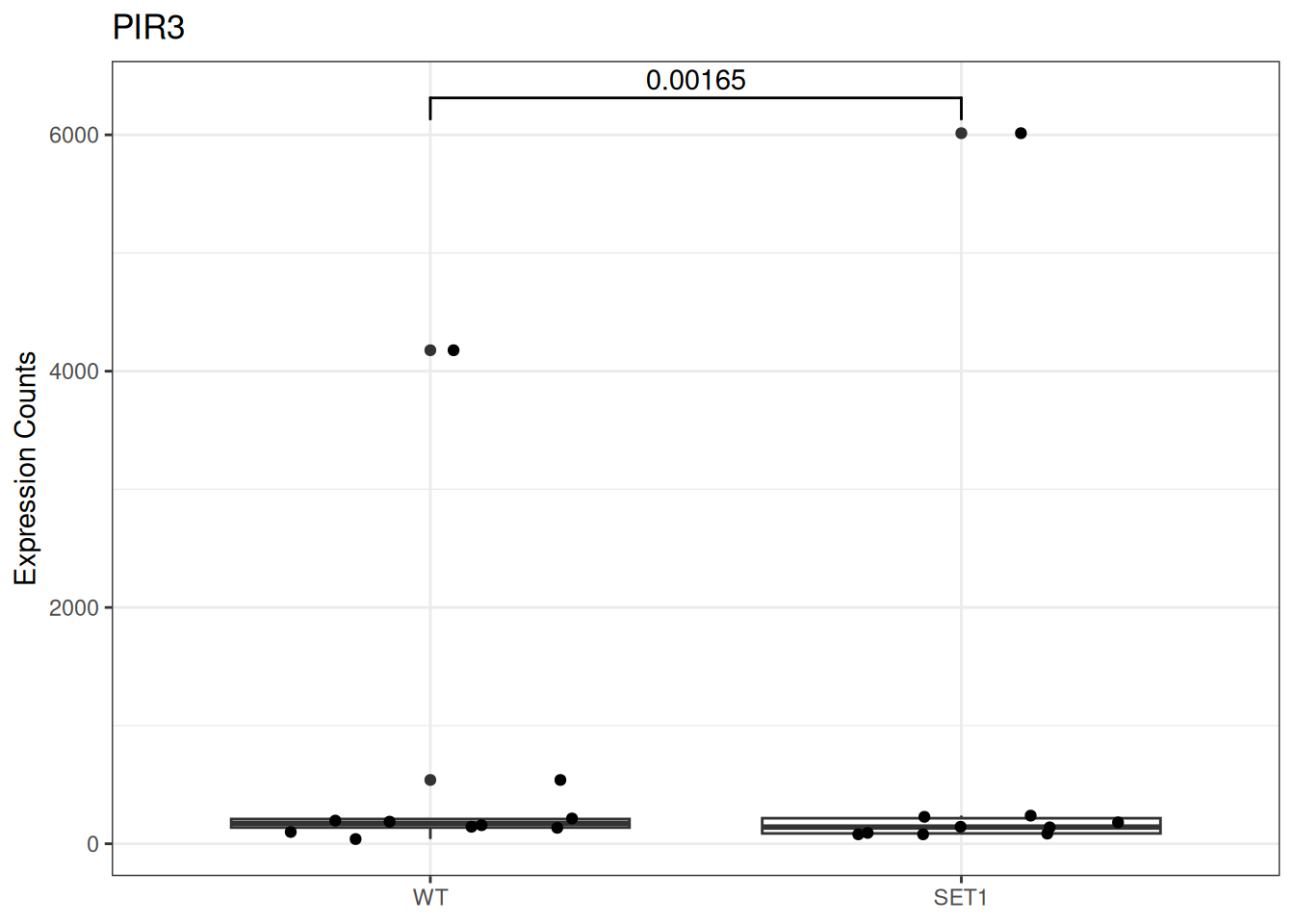
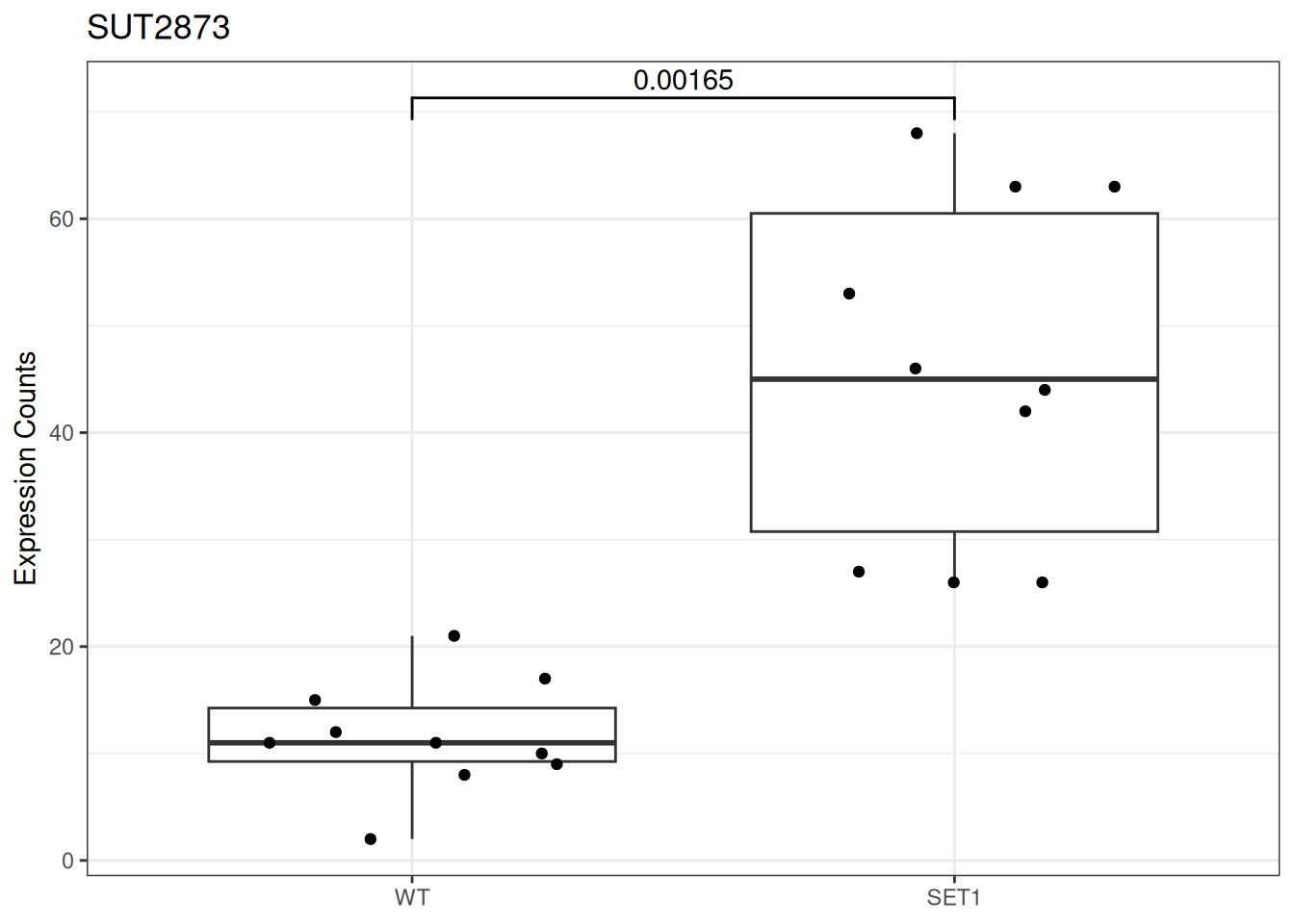
Based on differential gene expression analysis (SET1 vs. WT) results, draw the boxplot of for the top 3 genes with the smallest adjusted p-value, add individual data points on the boxplot and show p-value above the boxes with horizontal bar (with the help of the {ggsignif} pacakge).
Import the differential gene expression analysis (SET1 vs. WT) results file toy_DEanalysis.csv into RStudio and name it as de_res.
# Import DE resultsde_res <-read.csv("../exos_data/toy_DEanalysis.csv", header =TRUE)head(de_res)
Based on the counts data, build data frame for the LOH1 gene for the boxplot. This data frame should contain:
a column of counts for SET1 and WT samples,
and a column for corresponding the sample group.
Attention: In order to avoid hardcoding the gene name or sample name, use a variable instead.
# create a variable to store gene namemy_gene <-"LOH1"# even bettermy_gene <- target_genes[1]# create another variable for the number of sample1:10# old way
[1] 1 2 3 4 5 6 7 8 9 10
n_sample <-10seq(n_sample)
[1] 1 2 3 4 5 6 7 8 9 10
seq_len(n_sample) # safer, take only non negative number